Nghiên cứu cơ chế phản ứng nhiệt phân của gốc tự do furyl bằng phương pháp tính toán lượng tử
Từ khóa:
gốc tự do furyl, lignocellulose, năng lượng sinh học, nhiệt phânTóm tắt
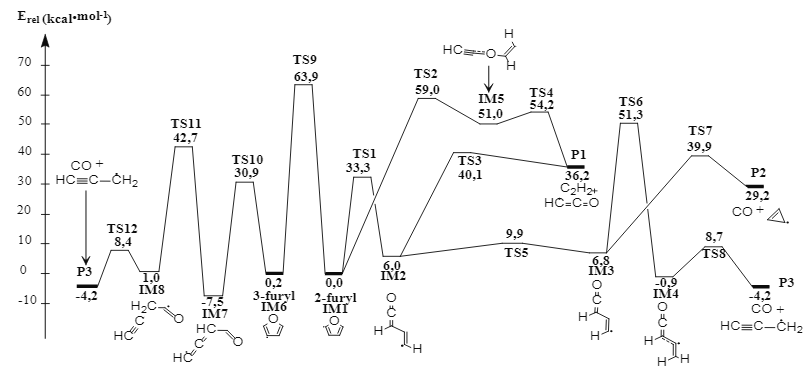
Trong nghiên cứu này, nhóm tác giả sử dụng phương pháp tính toán tổ hợp có độ chính xác cao CBS-QB3 để xây dựng cơ chế phản ứng nhiệt phân các gốc tự do của furan - thành phần quan trọng trong quá trình sản xuất nhiên liệu sinh học thế hệ hai từ lignocellulose/lignoxenluloza. Cơ chế phản ứng được làm rõ như sau: (i) Sự chuyển hóa giữa 2-furyl 3-furyl không thuận lợi do rào cản năng lượng lớn (63,9 kcal∙mol-1 ở 0 K); (ii) 2-furyl có thể bị nhiệt phân thành 3 kênh sản phẩm với thứ tự thuận lợi về mặt nhiệt động là C2 H2 & •HC=C=O (P1) > CO & c-C3 H3 • (P2) > CO & HC≡C‒CH2 • (P3); (iii) 3-furyl chỉ nhiệt phân theo duy nhất một kênh sản phẩm P3. Các giá trị tính toán của các thông số nhiệt động học (ví dụ như nhiệt tạo thành, entropy, nhiệt dung riêng) trong khoảng nhiệt độ từ 298 đến 2.000 K cho thấy sự tương đồng với các số liệu đã được công bố rời rạc trước đó. Do đó, các kết quả tính toán về cơ chế và nhiệt động học có thể được sử dụng cho các nghiên cứu sâu hơn về mô hình hóa và mô phỏng động học các hệ phản ứng liên quan đến gốc tự do furyl cũng như công nghệ chuyển hóa lignocellulose/lignoxenluloza
Chỉ số phân loại